Watch the video of Ed Wild’s address to the European Huntington’s Association annual meeting in September 2012, summarizing how therapies research works and explaining the most exciting ways researchers are trying to study and treat HD.
DR ED WILD: So, hello; good afternoon. My name is Ed, and the reason I have three different job descriptions on my title slide is because I am very bad at saying no to things. Particularly things to do with Huntington’s disease.
ED: So, to demystify, because that’s why I’m here, the top one means that I am a Huntington’s disease researcher. The middle one means that I’m a neurologist; and the bottom one means that I co-founded HDBuzz with my friend and colleague Jeff Carroll.
ED: What I’m here today to do is very easy, anyone could do it and it’s no problem at all. Simply to explain two and a half days of hardcore science in 50 minutes. So, I’m sure that won’t be a problem.
ED: Needless to say, what I will have to do is focus on the more exciting things, the things that are most likely to be of interest and relevance to you guys. Who are either living with Huntington’s or you know someone who is. Or you may be professionals who are involved in the care of Huntington’s disease patients, and families.
ED: So what I’ve decided to do, is boil it down. It’s been an amazing meeting; it’s been absolutely great. I’m going to try and boil it down to four areas. Hardcore science areas.
ED: The first one is about prevalence, how common is Huntington’s disease.
ED: The second is understanding the disease. Sometimes it may appear that we do a lot of staring down microscopes and describing Huntington’s disease, when what we really should be doing is treating Huntington’s disease. But of course you can’t really treat Huntington’s disease until you understand it. While we do clinical trials and drug trials on one end of the spectrum, at the same time it’s absolutely essential that we have basic research going on into understanding what causes the disease. How cells become damaged and so on. Because that’s how we keep our research pipeline full and always moving forwards. So some comments on that.
ED: Then stem cells I want to touch on specifically because I know that everyone wants to know about them, they were discussed at some length at this meeting.
ED: But of course really the thing that I guess you want to hear most of from the meeting is how are we doing with therapies and possible treatments. That we hope may slow down the progression of Huntington’s disease, or indeed provide new treatments for the symptoms of HD, to give people a better quality of life for longer. So the bulk of my talk is actually on therapies.
ED: Now I come to review this slide, I realise actually that I’m doing therapies as number three and stem cells as number four, so that was really just to make sure you were concentrating.
ED: But first, I need to do my characteristic plug for HDBuzz. Hands up if you have heard of HDBuzz. Excellent. That’s wonderful. You are clearly some of the best informed people in the world when it comes to Huntington’s disease. Not because you’ve heard of HDBuzz but because you’ve all made the effort to come here to this meeting. So I’m really pleased that you’ve heard of it. Hands up if you find it useful yourselves. Excellent, well, thank you very much.
ED: Actually I can recognise in the audience several people who represent individual European associations who very kindly donated money in support of HDBuzz. It’s our great pleasure to have that partnership and to be able to provide the research news. So I won’t need to spend very long plugging it. In fact what I really want to do is just draw your attention to some aspects – oh, there’s Jeff. Not sure if he’s in the gallery – no, he’s still around somewhere.
ED: I want to draw your attention to some aspects of HDBuzz that have been relevant to this meeting. Because we’ve been really putting in a lot of effort to try and bring this meeting alive and bring it to the global HD community through HDBuzz. So, any Twitter followers in the audience? Hey, excellent.
ED: So we’ve been tweeting research updates throughout the meeting, HDBuzzFeed is our Twitter handle. We’ve been doing that in quite some detail. We’ve also been providing updates on Facebook. Then at the end of each day we’ve been producing these summary news stories, which are pooled from the Twitter feed. They’re up online already if you go to hdbuzz.net, the English articles are up. As soon as they’re up, our army of translators begin translating them into our 12 languages. So, basically every single science session is covered on HDBuzz in those stories.
ED: So if there’s anything I miss today, you can always go back to HDBuzz and see what we said about it during the conference. The other thing we’ve been doing is the EuroBuzz feature which we were doing at the end of each day, interviewing scientists, having a bit of fun, explaining HD research. Those sessions will be available hopefully in the next week or so as two videos, which will be up on the HDBuzz site. We’ve specifically picked the scientists who are presenting the research that are most likely to be useful to you guys, the community members. We’ve helped the scientists to explain their research in terms that hopefully patients and family members will understand. So those will be up later in the week.
ED: Have I missed anything? No, I think that probably covers it. On the subject of translation, though, these are the 12 European languages that HDBuzz is now available in. So make a note of the country code that corresponds to your language. On the HDBuzz site you have to click on this little globe icon at the top of the page to get the dropdown list of languages.
ED: This is a very exclusive sneak preview of HDBuzz 2, which will be coming hopefully later in the year. It’s a redesigned site. A number of things should be a lot easier to navigate. But in particular the language dropdown box is a lot more prominent, so hopefully that will encourage a lot more participation from non-English speakers.
ED: The other thing we’re doing to try and help people who are brand new to HD research is this ‘start here’ feature. So if you or someone you know want to find out about Huntington’s disease research but hasn’t got a clue where to start. You can send them to HDBuzz and tell them to click on the ‘start here’ button. That will talk them through Huntington’s disease, tells them about us. Then we go straight into treatment research, starting with the most exciting things from the ground up, as we say. So that’s all pretty cool.
ED: The other thing is the new version of HDBuzz will allow you to download PDFs. So if any of you have relatives or friends who are interested in finding out about research but are not online, or don’t like using the internet or don’t use Twitter, or whatever, you can print off these PDFs. They’re basically individual factsheets, each one about a research topic. All in plain language. If you run a support group or if you’re a member of a support group you can print as many of them off as you’d like; it’s all completely free of charge. They’re in all the 12 languages that we have. The HD clinics that you go to, go along, take some factsheets with you. Maybe tell the clinic organisers that they can get these PDFs and put them in their clinics. So the more people that read our stuff the better.
ED: Okay, so before I go onto present some of the specific science that came from this meeting, because there were many people in the audience who didn’t understand a lot of what was said. Because ultimately each scientist can only work on a small area of Huntington’s disease. They very often will get on the stage and present as if everyone understands everything they already understand, and all they’re presenting is a few new things. But of course really we need to try and get everyone up to a certain level of understanding so that they can then appreciate what’s been said during the meeting.
ED: So I want to start with some basics about how research works, how we do science, and then we can move onto what’s new. So, I often talk about Huntington’s disease research and finding effective treatments for HD as being a bit like a voyage up a mountain. In particular, in the case of HD, it’s sort of like a mountain where we don’t even know how tall it is. So we don’t know how long it’ll take us to get there. All we know is it’s hard and it’s big.
ED: We’ve heard the word ‘hope’ already, and it’s a word that’s used a lot. But people sometimes tell me that they’re sick of hearing the word ‘hope’, because they’ve been hearing it for so long and still we don’t have treatments. But I do think that there is still room for hope in the lives of people living with HD. But what I would encourage people to have as well as this hope, hope that we will reach the top of the mountain behind that cloud, is a different, or rather special kind of hope. That I refer to as substantive hope.
ED: So if you were trying to climb this mountain, you’d be a bit bonkers – by which, sorry, I do have a tendency to use strange old-fashioned English words. You’d be a bit mad if you thought you could just set off one morning and get to the top of that mountain behind that cloud. That wouldn’t be a good way of going about it. So my view is that we need to break the journey down into small steps. That way we keep the mountain in the back of our minds, but if at any time all we’re worrying about is getting up that step and that step and that step, suddenly the journey to the top of the mountain is a lot more manageable and a lot less intimidating. We’re also much less likely to be disappointed if we wake up one step up instead of at the top of the mountain. So, this is what I refer to as substantive hope.
ED: What it means is that we need someone to fill in the steps, and hopefully that’s what I will do. That’s what we will continue to do through HDBuzz. But I don’t want you to lose this overarching sense of hope, so I’m going to give you five big reasons to have hope.
ED: I like to say Huntington’s is the most curable incurable brain disorder. Some people don’t like the word ‘cure’, some people don’t like the word ‘incurable’. I use them both very, very cautiously. I see that Charles is sitting there gently fuming. Here’s what I mean by that. Nearly 20 years ago the gene was discovered, and Huntington’s is unusual in that everyone with that mutation gets Huntington’s disease. Everyone with Huntington’s disease has the same basic mutation. That immediately gives us a head start over Alzheimer’s disease, Parkinson’s disease and motor neurone disease. We know exactly what we have to do to treat Huntington’s disease. We just have to get rid of the effects of this mutation.
ED: Lots of treatments have already worked in animal models of Huntington’s. So you can put the gene in a mouse or a fly or a Moomin, for any locals in the audience. You can give drugs or set treatments to those animals and the disease is less bad. So we know that it’s treatable in these animal models. All we need now is a pill that turns humans into mice, and then we’ll be fine.
ED: But seriously, we have come a long way. I know it feels like we haven’t, because the treatments aren’t there yet, but we really are making progress.
ED: Number two sounds a bit boring and is about the global infrastructure. Unlike many other conditions, we have fantastic organisations like EHDN, like the EHA, in America they have a Huntington’s study group. The patient groups are absolutely critical, and they’re extremely well organised in Huntington’s disease, because it’s a family disease and it’s a community disease. Because of the way that interacting with patients moves doctors and professionals, it’s very rare for a researcher or a professional to leave the world of Huntington’s disease. Because we want to help you guys. We’re essentially in for the long haul. So we’ve got this phenomenal worldwide infrastructure.
ED: In addition we have this thing call the CHDI Foundation. They don’t pay my salary, I don’t take any money from them. They’re a non-profit organisation, rather like a drug company, but they solely focus on drug treatments for Huntington’s disease. They’ve put an awful lot of money and organisation into that. I really do think that that strategy… If anything will succeed, I think that strategy of behaving like a drug company, like a Pfizer of a Glaxo, but focusing just on Huntington’s disease, is the one that will succeed.
ED: Talking of big drug companies, there are big multi-national, multi-billion dollar drug companies that are interested in treatments for Huntington’s disease. At this meeting alone we heard from representatives GlaxoSmithKline and Pfizer – huge drug companies. One thing they have is the money to run clinical trials, which is otherwise very difficult to come by. They also have the expertise to get drugs licensed and marketed. But I think really the fact that these big organisations is interested is a sign that the quality of Huntington’s disease research is very healthy. It’s like when sharks appear in a sea, it means that the sea is very clean. I’m not saying that they’re sharks, but they’re high level predators! OK.
ED: And crucially the whole world of Huntington’s disease and the global community’s expanding. In recent years we’ve seen the South American network, the Chinese network, and there’s networks popping up all over the world. They’re not isolated from each other. Essentially the world is becoming one big Huntington’s disease network. That’s a huge driver when it comes to research and driving forward work into treatments.
ED: Number three is something that we call the golden window of opportunity, and Sarah Tabrizi presented this slide. Essentially, you’re born without signs of the disease. If you have the gene that causes Huntington’s, at some point, unless we can come up with treatments that will delay onset, symptoms will develop. Before the symptoms begin, though, as many as 20 or 30 years before the symptoms begin, we know that the brain cells, the neurons, are sort of struggling. They’re not working perfectly, because of early, subtle effects of that mutation. However, because we can do a genetic test, we can predict who’s going to go on and develop those symptoms.
ED: So if we can develop treatments that will relieve some of those pressures on brain cells, we should be able to push forward the age of symptom onset. We have that in HD; they haven’t got that in Alzheimer’s or Parkinson’s, because they have no way of knowing who’s going to get the disease. That will help us when it comes to treating people in the future, but it also helps us now when it comes to studying the disease.
ED: I think my numbers might be out; I’ve gone from three to five. I’m not doing very well with numbers today. Having symptoms doesn’t mean it’s too late. So that slide was about preventing onset, what about if you already have symptoms when we develop treatments? Well, there’s a technique called gene silencing, which I’m going to talk about later. Essentially it means you could switch off the HD gene. It’s not possible in humans yet, but it’s been done in many mouse models. Essentially the mouse is born with the HD gene or the mutation, and at some point it becomes unwell. If you switch off the gene after the mouse has become unwell, the mouse gets better, the symptoms get better. If you look at the mouse brain under the microscope, the damage in the brain cells actually gets better as well.
ED: Now, none of this means that Huntington’s is not a neurodegenerative disease. Brain cells do die, and once they die there’s no bringing them back. But for any person, we’re optimistic if we can slow down the damage in those cells that people who already have symptoms may see improvements. Or at least may benefit from those treatments. So having symptoms doesn’t mean it’s too late.
ED: This is the last one – this is actually number five. Science is cumulative. Science is like this glacier. Snow falls at the top of it, and each snowflake doesn’t make much difference. But over years and decades and hundreds of years, the weight builds up. Eventually what you end up is this huge structure which can move mountains. To my mind, that’s how science works. This brings us back to these little steps of substantive hope. Each step takes us a little bit closer. Each day we know a bit more than we did the day before, and tomorrow we’ll know a bit more.
ED: Okay, very brief diversion to how drugs develop. Because this I think gives an idea of why it takes such a long time. This is what I’ve mentioned already, the drug development pipeline. Yesterday we heard about treatments that were all the way through this pipeline. Essentially what happens is you have to do a lot of work in cells and the lab, and messing around with chemicals, to put a drug into the pipeline. Once you have a drug, or a target and a drug that match each other, then you test it. You test it in cells, you test it in worms and so on and so on.
ED: Then once it’s been tested exhaustively, then you can start your human trials. Those are divided into several stages. So first you have to establish in healthy volunteers that the drug is safe. Then small numbers of patients for safety. Then the big trials where you find out whether the drug actually works in patients or not. It takes at least 10 years to get a drug through this pipeline, and the bottom bit alone often takes around 5 years. So we discovered the gene in ‘93, and that’s when really we were able to for the first time identify possible targets in cells.
ED: But there are targets that we’re just hearing about this year that are new and exciting, where those drugs will have to be developed from scratch. So I think for me the message number one is it does take a long time, but we are working on it. Number two, the pipeline is full. Right now for Huntington’s disease there are drugs at the very early stages and there are drugs that are right about to go into clinical trials. There are clinical trials going on now, make no mistake. There are drug trials happening now in Europe and in America of things that we hope will slow down Huntington’s.
ED: Okay, here comes the science part. Everybody still awake? Excellent. So the first thing I want to talk about is increased prevalence. So prevalence basically means how many people are there at any one time in a particular population who have Huntington’s disease? Michael Hayden spoke about this on the first day of the meeting. When we talk about prevalence we talk about numbers per 100,000 or per 10,000. What this means is that if you have 100,000 people in a population, 4 to 6 of them would have signs of Huntington’s disease. This is the traditional textbook figures, and it comes from studies that were conducted before or around the time the gene was first discovered.
ED: Now, a lot has happened since then. We’ve become a lot more aware of how to diagnose Huntington’s disease clinically. In other words, if Huntington’s is well known in a family, it’s fairly easy to pick up. Because someone says 'Oh, my mother had Huntington’s disease and I want to have the test.’ But new cases where the family history’s not known or where it’s emerging for the first time, it’s previously often taken two or three generations for those diagnoses to become apparent. But now I think we’re getting a bit better at picking them up.
ED: People are living longer as well, which enables the signs of later onset Huntington’s disease to emerge. I think a lot of what’s happening as well is that people are simply gradually lifting the stigma of the disease. So that people are more prepared to talk about it with their families and with their carers and doctors. So that essentially we’re just a lot more aware of cases out there.
ED: So the HD Association in the UK spoke to Sir Michael Rawlins who’s the Chairman of NICE, which is a health body in the UK. Said ‘We’re told that there’s 4 to 6 per 100,000. We actually have twice as many people as that already on our books that we’re caring for. So there’s no way that figure can be correct.’ So Michael Rawlins has now done a study where he’s looked at the nationwide general practice database, and has produced this figure of at least 12 per 100,000 for Huntington’s disease in the UK.
ED: Michael Hayden’s group in Canada did a similar study and have produced a figure of up to 15. So it’s looking like Huntington’s is at least twice as common in the population as we thought.
ED: This probably doesn’t come as a surprise to you guys, because this news basically came from the community. From families saying ‘There’s no way that Huntington’s is as rare as that.’ So really this is basically science vindicating what the community’s told us.
ED: What it means is that governments and health organisations need to dedicate more resources to caring for Huntington’s families and patients. It also means that in the future, as the population ages, we may end up with even more cases of Huntington’s disease. So that’s something to be aware of.
ED: But moving onto the lab science, as it were, talking about pathology and treatments. Just a little perspective, because I’m going to be talking about cells and genes and all that sort of stuff. This is an artist’s impression of our galaxy, the Milky Way. It contains 100,000m stars. If you multiply that number by 100 you get the number of cells in the human body. So it’s quite a lot. Each and every one of those cells contains our genome, all of our genes, including two copies of the huntingtin gene.
ED: The cell has a nucleus where the DNA is, and a cytoplasm where all the other stuff happens. Cells are basically powered and all the business of the cell is done by these machines called proteins. In the nucleus is our DNA, our genes made of DNA. One gene is a recipe for one protein. On the way to making the protein this message molecule, called RNA, is manufactured. Then it’s used to make a string of these little blobs called amino acids, and those then assemble into a protein. In the case of Huntington’s disease, the huntingtin gene leads to huntingtin RNA, and that then leads to the huntingtin protein.
ED: As you know, huntingtin genetics, it’s all about CAG. We have this repeat of CAG, these letters in our gene. Each CAG corresponds to one little building block called glutamine, which scientists call the letter Q. What that does is it takes this normal huntingtin protein, which looks something like that. The CAG bit is here, or rather all those little Qs, the glutamines are there. Having too many glutamines in a row changes the shape of that, but it also changes the shape of the whole protein.
ED: So the whole protein changes in shape as a result of that small lengthening of this glutamine bit. If you’re a protein then changing your shape will change your function. Because the functioning of proteins depends on their shape.
ED: Which leads to public enemy number one, which is this, the mutant huntingtin protein. This is a microscope picture of the huntingtin protein. As you can see, it forms into these mountain-like blobs called aggregates. But we think the poisonous bit is actually this bit here, before it forms into the aggregate. So this is the cause of all of the problems in Huntington’s disease, the mutant huntingtin protein. It does loads of stuff. It basically messes up loads of things that need to happen for our cells to be happy. Our neurons, unfortunately, suffer as a result. So that’s my Huntington’s disease basics 101.
ED: What have we learnt during this meeting about how that protein causes harm? Well, for me there were two striking talks.
ED: This is Gill Bates from London, and she studies a protein called HDAC 4. Now, HDACs are a family of enzymes which control which genes are switched on and off. HDAC 4 has long been of interest when it comes to a possible treatment for Huntington’s disease, because the huntingtin protein messes up the switching on and off of other genes. But Jill has been studying all of these HDAC enzymes with a particular emphasis on HDAC What she’s uncovered is that it seems the effects of HDAC that protect – or that are to do with Huntington’s disease – are actually happening not in the nucleus where the DNA is, but in the cytoplasm, where there isn’t any DNA. So this is a major upheaval in our understanding of this important protein. Jill’s lab is now looking into why that might happen.
ED: The other talk that really fascinated me on the basic science of Huntington’s was by Ray Truant who has been looking at the structure of the protein and how it moves around in cells. He describes the protein as being a bit like a slinky, like a spring. It has lots of these stretchy bits and it changes shape. It may have something to do with sending signals around the cell based on what shape it has.
ED: In particular, Huntington’s disease has something to do with the way our cells handle stress. The polyglutamine that all of those extra glutamines in the mutant huntingtin protein in some ways seem to be making the huntingtin protein less able to help the cell deal with stress. Something about that glutamine stretch makes the huntingtin protein less springy. Ray calls it his ‘rusty hinge hypothesis’, which is quite useful. We actually interviewed Ray Truant on stage, so once the EuroBuzz videos are online at HDBuzz, you can hear Ray talking directly about his science. In terms that are pretty easy to understand, I think.
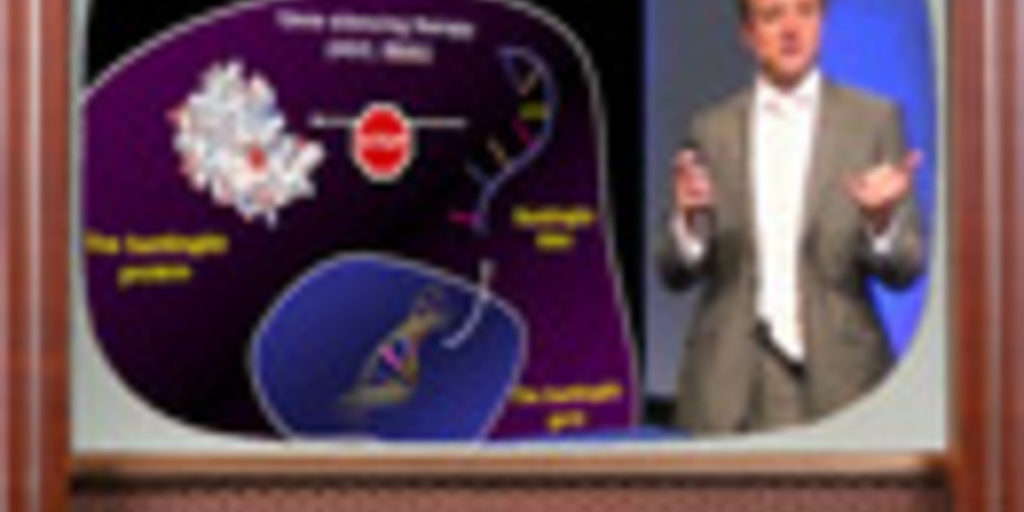
ED: Moving on, then, to therapies. How long have I got left? Some minutes, excellent. I’ll keep going until there aren’t some minutes. So, gene silencing therapy; hands up if you’ve heard of it. I did mention it earlier so you should all have your hand up, but that wasn’t what I was asking. This is one of the most famous possible approaches. Basically, if your house is flooding, it’s important to mop up the water. But what’s most important is turning off the tap. Or faucet, if you’re an American-English speaker.
ED: What gene silencing or huntingtin lowering therapy basically involves is admitting that it’s very difficult to change the DNA in all of our cells. But saying ‘Well, maybe if the huntingtin RNA gets made but we can stop that from getting turned into a protein, then there won’t be as much of the mutant protein hanging around.’
ED: It turns out that our cells actually have mechanisms already for getting rid of unwanted RNA. So, all that’s needed really is to design drugs that stick to the huntingtin RNA and say to the cell ‘Excuse me, get rid of this. What I’m attached to, get rid of it.’
ED: It works, essentially. It’s been tested now in a number of cell models and mouse models of Huntington’s disease. There are a number of different approaches to making these molecules, and the number of ways of getting them into the brain of the animal. But basically, every time these huntingtin lowering approaches have been tried, they have worked in that animal.
ED: There’s a trial of this going on right now in motor neurone disease, also known as ALS, in humans. In the past 12 months – and this was discussed at this meeting – we now have three different trials done in monkeys, which have large brains quite similar to human brains, showing that the drug… You can get the drug into the brain where it’s needed, and where the drug lands, the gene is switched off. Those monkeys did not, on the whole, experience bad side effects from this. This is really happening, guys.
ED: So, in the next 12 to 18 months there’ll be at least one trial of gene silencing, the very first in Huntington’s disease in human patients. The first trials will probably happen in America and they’ll probably involve patients with early to moderate Huntington’s disease. But very rapidly I would expect there to be trials happening in Europe as well.
ED: We’ve written about this a lot on HDBuzz, and these are three articles that you might want to look at. So all you do is type hdbuzz.net/ then either 69 or 58 or 23. Once you’re in one gene silencing article you can easily click through to others.
ED: A lot of people, whenever I speak about gene silencing or huntingtin lowering, they want to know how it works and how it will actually be in practice. Right now the way it works is that these are drugs that are made of DNA and made of RNA. If you take them as a pill they just get destroyed by the acid in the stomach, they don’t get to the brain.
ED: So right now that means treatments that all have to be injected into the nervous system. Either into the spinal fluid at the base of the spine, or by directly being pumped into the brain or the ventricles that are in the middle of the brain. So, every silver lining has a cloud. These are extremely promising drugs, but as they are envisaged now, they will involve at least injections into the spine, possibly injections into the brain. But if it works, it will be worth it.
ED: Then there’s the question of whether we should turn off one gene or both copies of the gene. So everyone has two copies, and most people with Huntington’s have one normal and one abnormal copy. If we could turn off the one that’s making the mutant protein while leaving the other one still producing the healthy protein, that might be better. That’s something that’s being worked on as well.
ED: Bev Davidson was the person who presented this huntingtin lowering research, and yesterday evening we interviewed her for EuroBuzz. So, day two of our EuroBuzz video contains a really nice interview with Bev, who’s one of the leading lights in the field of huntingtin lowering. A very funny woman as well, which is always important.
ED: So, a brief word about what we call post-translational modification. It’s total jargon. What it basically means is that once a protein’s made in cells, that protein might need to go to different bits of the cell. It’s a bit like delivering a parcel. You stick a label on it like a barcode or an address label on it, and that’s what cells do, basically.
ED: One of those little tags or labels that’s attached to cells is called acetyl. Doesn’t matter what it is or what it’s called. But basically there’s a tag that tells the cell to move the protein into this bag of enzymes which dissolve the protein and get rid of it. That is how the cell gets rid of big proteins like the huntingtin protein.
ED: There’s an enzyme called sirtuin 1. One of the things that that enzyme does is it gets rid of these acetyl tags. So, if you follow me, what that enzyme does is it tells the cells not to get rid of the protein. So we wanted to encourage them to get rid of the bad protein. So what we want to do is decrease the activity of that enzyme. There’s a lot of going backwards and forwards here.
ED: Bottom line is that if you can come up with a drug that inhibits this sirtuin 1 enzyme, you should be able to tag these proteins and tell the cell to get rid of them. Siena Biotech – Andrea Caricasole is the guy who presented yesterday about this drug – has developed an inhibitor of this drug called Selisistat – difficult to say with a dry mouth. Selisistat. This enzyme, when he looks at it in the lab and in mouse models for Huntington’s, encourages the tagging of that protein with that acetyl tag. Which encourages the removal of the protein.
ED: This drug has had a number of beneficial effects. We don’t know whether it will work in humans, but this is a trial that’s going on right now. It’s called the Paddington Study in part. It’s supported by the EU and by the Euro HD network. The phase 1b trial completed, Phase 2a just finished last week in Europe. So far the drug appears to be safe. So, the results of that trial have yet to be analysed. The next step, if they look good, would be to move to a larger trial to see if it can actually slow the disease.
ED: A brief word on synapses, because I want to move on now to something that’s really cool called phosphodiesterase inhibition. So as you probably know, signals in the brain are electrical. Our brain cells transmit signals electrically. But at the end of the brain cell there’s a gap. You can see it here. A gap between the two brain cells, and that’s called the synapse. The electrical signal reaches the end, but the signal gets across the synapse in chemical form. So what that means is that in the second cell it receives a chemical message, and then it has to convert that to an electrical message for the transmission to take place.
ED: So, this is the chemical outside of the second neuron, and this is the neuron. Basically, the chemical enters and then a cascade of signalling happens. So one little thing triggers a big cascade. Then in the process of cleaning up that message these enzymes called phosphodiesterase, or PDE enzymes, come into play. They basically sweep up all of these signalling molecules. Now, in Huntington’s disease, what’s been observed is that the synapses, the connections between the brain cells, work less effectively. One of the reasons may be that these PDE enzymes are overactive, or they’re cleaning up all of these signalling molecules too early or too much, which makes the signalling less efficient. So, one of the aims – and this has come out quite recently, just in the past two or three years. There’s been a lot of excitement about whether we could inhibit these PDE enzymes with a drug, and whether that might improve the functioning of synapses.
ED: This is where the drug giants come in, Pfizer and Glaxo, GSK. Pfizer has a drug with this extremely catchy name, that inhibits a PDE called PDE10. Restores some of the synaptic problems in HD, and improves damage to cells in HD mice. They’re working with CHDI to test these cells. I have to say that the results that were presented look really encouraging. This is a drug that really does seem to be making a difference to the functioning of synapses. If we’re lucky it may have more long-standing effects as well. Pfizer’s a real drug professional. It has an extremely sensible plan for first looking at patients to see whether we find the same signalling problems and then moving forward into a human trial. This is all happening now and it’s likely to be… The human studies will be happening next year with a bit of luck.
ED: And then GSK also has a PDE programme. The one they’ve chosen to focus on is PDE4. That’s the name of their drug. You can’t buy it online, unfortunately, even if you can remember the name. Their drug has also shown promising signalling improvements when they test it in cells in a dish. In particular they’ve noticed that some of the electrical clues that tell us whether the cells would be good at learning new things seem to be improved in the presence of their drug. They’ve tested it now in healthy volunteers. There were some concerns about side effects like nausea in healthy volunteers. But they nonetheless plan to move forward soon into human trials to see whether overall this helps or makes things worse. We’d be optimistic that it’ll help.
ED: Because of the excitement, we wrote a Buzz article about PDE inhibitors, which is buzz.net/86. Okay, we’re getting there. Oh, this is my impression of Pfizer and Glaxo. [Laughter]
ED: We’re now at a stage where we basically have these two elite athletes racing towards trying to produce drugs for Huntington’s disease. So it’s a pretty healthy situation to be in.
ED: This was intriguing to me; this is the last drug that I’m going to talk about. Buproprion. This was something new that I learned at this meeting. Bupropion is also sold as Wellbutrin. It’s a drug to help stop people smoking. But apathy is what it’s being aimed at in Huntington’s disease. Apathy is when patients with Huntington’s disease have trouble motivating themselves to get out of bed or to go to work, or to go out and socialise. Sound familiar? Yes. I hear about it a lot. It’s something we really struggle to treat. So it would be great if this works. The study’s called Action! HD and it’s happening in Germany; it’s enrolling now.
ED: So that was pretty exciting news, and we look forward to hearing what the outcome of that is.
ED: So the final thing, assuming I still have some minutes… Two, okay, [Laughter]. The final thing, then, is stem cells. Big headline grabbing things, stem cells. A stem cell is a cell that can turn into any other cell type. So they sound amazing, and they are. We all began as stem cells. What stem cells are not is a magic treatment for Huntington’s disease. So you can’t just take stem cells, turn them into brain cells, inject them into brains and assume that brain cells that died will be replaced. Unfortunately it doesn’t work like that. It’s a bit like running lightning into a dead body and expecting it to wake up. It would be nice, but it’s not what happens.
ED: What they are right now, though, stem cells, is an extremely useful tool for studying Huntington’s disease. In particular these fellows called IPS cells, which stands for Induced pluripotent Stem cells. Basically you take a skin sample from a patient with HD, or with a mutation. From it, you turn the cells backwards into stem cells. Then you can turn them forward into neurons.
ED: Now, until now, the only way we could get living neurons that were human was either by buying them from a shop, an online store. In which case they’re nothing to do with Huntington’s disease and nothing to do with real patients. Or to use animal neurons. So you can take an HD animal and extract the brain cells from it. But what you can’t do is stick your hand into patients' brains, take out the brain cells, look at them under the microscope and test drugs on them.
ED: What IPS cells are enabling us to do now for the first time ever is to have actual cells that contain the real DNA of real patients, but behave and look exactly like neurons. So we can test drugs on them. We can look at them in incredible detail and say ‘What’s going wrong in these neurons?’ This really is an incredible advance. Believe it or not, this year and last year is the first time it’s been possible. It’s thanks to the big multinational stem cell consortium which involves people from all over the world.
ED: So Lisa Ellerby was the person who spoke to us about that, as well as Leslie Thompson, from California. So, Lisa was at great pains to point out that this is not a treatment yet. It may be, but we’re talking at least a decade, probably more. But what they are right now, definitely, 100%, is a really valuable research tool for studying Huntington’s and testing these drugs. Because it’s exciting – this is Lisa Ellerby by the way, who confessed that as well as pool, she was a big fan of wearing tutus.
ED: On day two of our EuroBuzz we interviewed Lisa as well, so do take a look at that video, because Lisa’s really good at explaining what stem cells are, what she did and how these will be useful research tools.
ED: I believe that is all I have to say. Thank you for giving me some extra minutes, and thank you for your attention. [Applause]





